Association¶
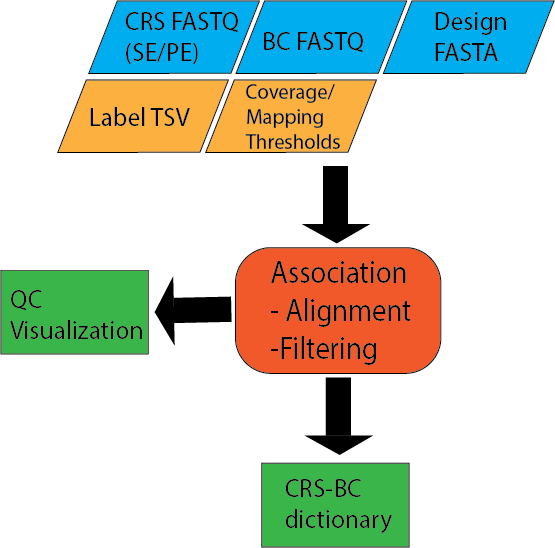
Input files¶
Fastq Files¶
2-3 Fastq files from library association sequencing –Candidate regulatory sequence (CRS) sequencing, 1 forawrd read and an optional reverse read if paired end sequencing was used –Barcode sequence, 1 read covering the barcode
Design File¶
Fasta file of of CRS sequences with unique headers describing each tested sequence
Example file:
>CRS1
GACGGGAACGTTTGAGCGAGATCGAGGATAGGAGGAGCGGA
>CRS2
GGGCTCTCTTATATTAAGGGGGTGTGTGAACGCTCGCGATT
>CRS3
GGCGCGCTTTTTCGAAGAAACCCGCCGGAGAATATAAGGGA
>CRS4
TTAGACCGCCCTTTACCCCGAGAAAACTCAGCTACACACTC
Label File (Optional)¶
Tab separated file (TSV) of desired labels for each tested sequence
Example file:
CRS1 Positive_Control
CRS2 Negative_Control
CRS3 Test
CRS4 Positive_Control
Note
If you provide a label file, the first column of the label file must exactly match the FASTA file or the files will not merge properly in the pipeline.
association.nf¶
Options¶
With --help or --h you can see the help message.
- Mandatory arguments:
--fastq-insert Full path to library association fastq for insert (must be surrounded with quotes) --fastq-bc Full path to library association fastq for bc (must be surrounded with quotes) --design Full path to fasta of ordered oligo sequences (must be surrounded with quotes) --name Name of the association. Files will be named after this. - Optional:
--fastq-insertPE Full path to library association fastq for read2 if the library is paired end (must be surrounded with quotes) --min-cov minimum coverage of bc to count it (default 3) --min-frac minimum fraction of bc map to single insert (default 0.5) --mapq map quality (default 30) --baseq base quality (default 30) --cigar require exact match ex: 200M (default none) --outdir The output directory where the results will be saved and what will be used as a prefix (default outs) --split Number read entries per fastq chunk for faster processing (default: 2000000) --labels tsv with the oligo pool fasta and a group label (ex: positive_control) if no labels desired a file will be automatically generated
Processes¶
Processes run by nextflow in the Association Utility. Some Processes will be run only if certain options used and are marked below.
- count_bc or count_bc_nolab (if no label file is provided)
- Removes any illegal characters (defined by Piccard) in the label file and design file. Counts the number of reads in the fastq file.
- create_BWA_ref
- Creates a BWA reference based on the design file
- PE_merge (if paired end fastq files provided)
- Merges the forward and reverse reads covering the CRS using fastq-join
- align_BWA_PE or align_BWA_S (if single end mode)
- Uses BWA to align the CRS fastq files to the reference created from the Design File. This will be done for each fastq file chunk based on the split option.
- collect_chunks
- merges all bamfiles from each separate alignment
- map_element_barcodes
- Assign barcodes to CRS and filters barcodes by user defined parameters for coverage and mapping percentage
- filter_barcodes
- Visualize results
Output¶
The output can be found in the folder defined by the option --outdir. It is structured in folders of the condition as
Files¶
- count_fastq.txt
- number of barcode reads
- count_merged.txt
- number of aligned CRS reads
- design_rmIllegalChars.fa
- Design file with illegal characters removed
- label_rmIllegalChars.txt
- Label file with illegal characters removed
- s_merged.bam
- sorted bamfile for CRS alignment
- ${name}_coords_to_barcodes.pickle
- pickle file containing a python dictionary of CRS/barcode mappings
- *.png
- Visualization of number of barcodes mapping to enhancers